
Chapter 55: Ischemia
Pathophysiology
Cerebral ischemia occurs when blood flow to the brain is reduced in either a global or a focal fashion. Given a sufficient degree and duration of either mode of deficit, irreversible changes occur that lead to cell death. A global reduction in cerebral blood flow generally results from extracranial events, such as diminished cardiac output from myocardial damage or dysrhythmia, severe hypovolemia, or loss of peripheral vascular resistance. When cerebral perfusion decreases in this generalized manner, compensatory collateral pathways themselves are affected and unavailable; thus the insult is more acute and there is less chance of reversal. More often, cerebral ischemia is focal and occurs in the distribution of one of the three major vascular territories. A localized reduction in cerebral perfusion often results from a combination of factors, including the disease state of the vasculature coupled with the individual's vascular anatomy and its autoregulation; the oxygen-carrying capacity, viscosity, and coagulability of the circulating blood; and cardiac output.
Once cerebral perfusion drops below a critical threshold (10 to 22 ml/100 g/min), a cascade of biochemical mechanisms is triggered within the ischemic tissue, which results in increased intracellular osmolarity.32 A rapid influx of water from the extracellular compartment causes the cell to swell, termed cytotoxic edema.17,47 In addition, a paralysis of cellular function ensues, leading initially to deterioration of the neuronal cytoplasmic membranes and eventually to the loss of vascular endothelial integrity. If reperfusion of the ischemic area then takes place, leakage of macromolecules and water into the extravascular space occurs, termed vasogenic edema.17,47 Cytotoxic edema begins almost immediately and continues for approximately 30 min to 1 hr after the ischemic insult; vasogenic edema begins to develop after 6 hr.
In general an ischemic focus has two zones—a central zone of infarction and the penumbra, or border zone. The central zone is where the cerebral blood flow falls most dramatically, resulting in infarction. The central zone is surrounded by the marginally perfused penumbra. The penumbra depends on collateral circulation for viability and may recover or necrose. In both the central and penumbral zones, neuronal viability is time dependent. The more profound the degree of ischemia and the longer the duration, the greater the irreversible damage. Electrical activity in cerebral tissues ceases at flow rates below 16 to 18 ml/100 g/min.20 Although electrically silent, cerebral tissue remains viable until cerebral blood flow (CBF) falls below 10 to 12 ml/100 g/min.6 In primates, when blood flow is reduced below this critical level, irreversible infarction occurs after only 1 hour.69 Lesser degrees of ischemia can be tolerated for several hours before irreversible infarction occurs. Prompt restoration of flow may prevent infarction, but the timing is critical because reperfusion after a certain period of time may actually be harmful; it can result in increased edema formation and compression of still-patent capillaries, propagating ischemia. Experimental animal models show that the “window of opportunity” appears to be species dependent. Therefore any single experimental model may not truly reflect the human in vivo situation. Also, the in vivo situation may vary from patient to patient.
Although it is unlikely that the central core of the infarct can be salvaged, the degree to which it can be minimized and the penumbra reclaimed will dictate the patient's prognosis. In the normal brain the capillary lumen closing pressure is only about 30 to 35 mm Hg.67 However, the closing pressure in poorly perfused brain may be even less. Research studies indicate that tissue hydrostatic pressure can rise up to 8 mm Hg as a result of swollen, ischemic brain cells.67 Furthermore, evidence suggests that mechanical compression of the capillary bed by the swollen brain cells occurs before the breakdown of the blood-brain barrier (BBB) when vasogenic edema develops.
A number of classic experimental models have helped shape the current theory that explains the development of cerebral ischemia.12,52,64,67,110,113 Three major pathophysiological derangements occur and cause the damage as a result of ischemia—a disturbance in cellular calcium homeostasis, regional acidosis, and free radical production.113
Calcium plays many essential roles in neuronal cellular physiology, which is dependent on a strictly maintained 10,000:1 concentration gradient between extracellular and intracellular calcium. Even small alterations of this gradient can dramatically affect neuronal physiology. Initially, with ischemia, energy failure causes depolarization of the cell membrane, release of neurotransmitters, and a consequent rise in intracellular calcium. The massive release of glutamate and other excitatory amino acids enhances the calcium influx by opening calcium channels. Calcium causes structural damage to the cell as a result of overactivation of lipases, proteases, and endonucleases, which attack cellular membranes and neurofilaments. Furthermore, neuronal hyperexcitability could be yet another detrimental effect of increased calcium.
Acidosis develops in densely ischemic zones partly as a result of free fatty acid formation caused by phospholipase-activated membrane breakdown. Acidosis also results from the loss of aerobic glycolysis, the most efficient way the cell has to produce adenosine triphosphate (ATP) from glucose, and anaerobic glycolysis supervenes. This later metabolic pathway produces much less ATP per molecule of glucose and yields an excess amount of lactic acid. Acidosis also promotes edema formation by the accumulation of intracellular Na+ and Cl– (with the obligatory accompaniment of water) by induction of Na+-H+ and Cl–-HCO3– antiporter mechanisms in an attempt to maintain intracellular pH. There is also excess intracellular sodium accumulation as a result of failure of the Na+-K+ pump caused by diminished ATP production. In addition to the promotion of edema formation, acidosis severely limits and inhibits recovery of mitochondrial function, which results in further decline of ATP production.
Free radicals are highly reactive and inherently toxic molecules produced in all aerobic cells. Cells possess both enzymatic and nonenzymatic defense systems that function as free radical scavengers. Anaerobic-aerobic transitions enhance free radical production. Therefore, when ischemia is followed by reperfusion, free radical production is heightened as a result of reoxidation of the reduced compounds that accumulated during ischemia. The microvasculature of the central nervous system (CNS) appears to be one of the major targets of free radicals, resulting in dysfunction of normal BBB and vascular regulatory tone, contributing further damage to the ischemic zone.
The maximum amount of postischemic edema occurs approximately 2 to 4 days after the ictus as a result of endothelial cell damage leading to significant breakdown of the BBB, which allows leakage of intravascular elements into the brain parenchyma. Early reperfusion and hyperemia (luxury perfusion) develop spontaneously in regions of acute infarction. This early hyperemia has been associated with pronounced edema.62 Vasogenic edema may produce sufficient mass effect to cause the patient's demise. Most current medical management is aimed at this phase of infarction. Unfortunately, despite such management, approximately 25% to 35% of patients admitted to the hospital with major territorial infarcts deteriorate.
The reparative process begins as early as 24 to 48 hours after infarction, when microglial cells proliferate around the periphery of the infarct and begin to phagocytize the necrotic tissue. As the cellular debris is removed, capillaries begin to form about the periphery of the infarction. These new capillaries do not have an intact BBB (thus allowing leakage of contrast material). Over time, the residual tissue becomes soft (encephalomalacia) as a result of atrophy and reabsorption of necrotic tissue, and it shows increased water content. If the reabsorption of necrotic tissue is complete, the result is a fluid-filled cystic cavity (macrocystic encephalomalacia17). Rarely, dystrophic calcification of infarcted brain may occur.99 This entire process is generally complete by 6 weeks; however, in some cases active reabsorption of tissue may persist for months.84
A substantial percentage of ischemic infarcts are further complicated by secondary hemorrhage. These hemorrhages commonly appear as small, patchy, petechial bleeds occurring 2 or more weeks after the infarction and are generally clinically inconsequential (Figure 55-1).63 However, individuals with proximal emboli causing large distal infarcts are more likely to develop early secondary hemorrhage caused by thrombolysis and reintroduction of high systemic blood pressure into the region of the damaged vascular bed (Figure 55-2).16 Up to 30% of patients with proximal middle cerebral artery (MCA) occlusion suffer secondary hemorrhage, usually within the first 4 to 5 days after the event.63 Thus early anticoagulation therapy is often withinfd in such patients during the week after ictus.
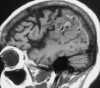 |
| Figure 55-1 Subacute infarct complicated by petechial hemorrhage. T1-weighted sagittal image shows gyriform pattern of high signal intensity in the cortical mantle (arrows), indicating T1-shortening from methemoglobin as a result of microhemorrhage. |
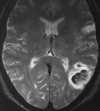 |
| Figure 55-2 Acute infarction with secondary hemorrhage on day 2 in a young woman who was grossly overanticoagulated. T2-weighted axial image shows partial hemorrhagic transformation of a left MCA territorial infarction. |
~ Previous ~ Next ~
~ Back to Chapter Index ~
~ Back to Magnetic Resonance Imaging main page ~